

VASCULITIS

ÍNDICE
1. GENERALIDADES Y CONCEPTOS BÁSICOS
2. FISIOPATOLOGÍA Y CLASIFICACIÓN
4. DIAGNÓSTICO
6. TRATAMIENTO
7. VASCULITIS DE GRANDES VASOS
8. VASCULITIS DE VASOS MEDIANOS
9. VASCULITIS DE VASOS PEQUEÑOS
10. ANEXOS
¡Bienvenidxs!, soy Interneuronamed y te dejaré los puntos clave para comprender a las vasculitis.
Sígueme en mis redes sociales (Twitter, Facebook, YouTube, Instagram) como @Interneuronamed, donde encontrarás resúmenes de medicina y más cosas que interesarán.
1. GENERALIDADES Y CONCEPTOS BÁSICOS
Vasculitis: inflamación y posterior necrosis de los vasos sanguíneos. La lesión endotelial conduce a trombosis y deterioro del flujo sanguíneo causando isquemia/infarto de los tejidos teniendo como consecuencia: ateroesclerosis secundaria acelerada del vaso afectado, lo que contribuye a la morbilidad y mortalidad.
Las vasculitis constituyen trastornos clínico-patológicos caracterizados por inflamación y daño a los vasos sanguíneos (lesión al interior del vaso + isquemia del tejido); puede circunscribirse a un solo órgano, como la piel, o afectar simultáneamente varios sistemas u órganos.
Síndrome vasculítico: son trastornos autoinmunes caracterizados por inflamación de los vasos sanguíneos, que pueden afectar a casi cualquier sistema/órgano y a meno conducen a una morbilidad significativa.
TIP INTERNEURONA: un vaso inflamado condiciona trombosis, embolismo, rotura de vaso, hemólisis, aneurisma, estenosis; y estos son algunas de las complicaciones de los pacientes.
Las vasculitis son enfermedades poco frecuentes. Las dos variantes que se diagnostican con más frecuencia son la vasculitis leucocitoclástica cutánea (lesiones dermatológicas persistentes) y la arteritis de la temporal o de células gigantes que llevan a urgencias al paciente (cefalea temporal unilateral). Las vasculitis tienen mayor frecuencia en mujeres que en hombre (autoinmunitario). En la infancia, la vasculitis más frecuente es la púrpura de Schonlein-Henoch (75% <7 años), mientras que la enfermedad de Kawasaki (faringitis, conjuntivitis, fiebre, lengua en fresa, compromiso cardíaco, ojos rojos, etc) es prácticamente exclusiva de niños <5 años, sobre todo hombres que puede complicarse por aneurismas de las arterias coronarias epicárdicas condicionando infartos a temprana edad. La arteritis de Takayasu (la enfermedad sin pulsos por la estrechez que causa) predomina en mujeres en edad fértil, mientras que la arteritis temporal en la tercera edad.
 |
| Arteritis de la temporal |
 |
| Vasculitis leucocitoclástica cutánea |
Las biopsias de tejido (estándar de oro diagnóstico) o los estudios angiográficos suelen ser necesarios para el diagnóstico. La agresividad del TX debe ser acorde con el grado de afectación del órgano terminal.
2. FISIOPATOLOGÍA Y CLASIFICACIÓN
En la mayoría de los síndromes vasculíticos son mediados, en parte, por mecanismos inmunopatógenos que aparecen en respuesta a algunos estímulos antigénicos. Es posible que intervengan factores como la predisposición genética, exposiciones y contactos ambientales, mecanismos regulares inmunitarios a algunos antígenos, entre otros.
 |
| Clasificación de las vasculitis |
Mecanismos potenciales de daño en las vasculitis primarias:
a) Formación de complejos inmunes: el principal y más ampliamente aceptado. Común de la poliarteritis nodosa asociada a hepatitis B, Púrpura de Henoch-Schönlein, vasculitis crioglobulinémica esencial.
b) producción de anticuerpos anti-citoplasma de neutrófilo (ANCA): común de la granulomatosis con poliangitis (granulomatosis de Wegener), poliangitis microscópica, síndrome de Churg Strauss. Aquí pueden elevarse cANCA (citoplasmático en los gránulos azurófilos contra proteinasa-3) o pANCA (perinuclear contra enzima mieloperoxidasa). Estos anticuerpos se pueden detectar por inmunoinfluorescencia o ELISA (más específico).
Otras patologías con ANCA: artritis reumatoide, LES, otras vasculitis, colitis ulcerosa, colangitis esclerosante primaria, hepatitis autoinmunitaria, fibrosis quística, enfermedades infecciosas y neoplasias.
c) Respuesta medidada por linfocitos T y formación de granulomas: común de las arteritis de células gigantes, arteritis de Takayasu, granulomatosis con poliangitis, síndrome de Churg Strauss.
 |
| Harrison 20a edición (2019) |
Clasificación del consenso de Chapel-Hill 2012:
Vasculitis de vasos grandes: Arteritis de Takayasu, arteritis de células grandes.
Vasculitis de vasos medianos: Poliarteritis nodosa, enfermedad de Kawasaki.
Vasculitis de vasos pequeños: Vasculitis asociada a anticuerpos anticitoplasma de neutrófilos (ANCA), polangitis microscópica, granulomatosis con poliangitis (Wegener), granulomatosis eosinofílica con poliangitis (Churg-Strauss), vasculitis mediada por inmunocomplejos (por anticuerpos antimembrana basal [anti-GBM], crioglobulinémica, Schönlein-Henoch [vasculitis IgA], urticaria vasculítica hipocomplementémica).
Vasculitis de vasos variables: Enfermedad de Behcet, síndrome de Cogan.
Vasculitis de órgano aislado: angitis cutánea leucocitoclástica, arteritis cutánea, vasculitis primaria del SNC, aortitis aislada.
Vasculitis asociadas a enfermedad sistémica: vasculitis lúpica, vasculitis reumatoide, vasculitis sarcoide.
Vasculitis asociadas con probable etiología: Hepatitis C, hepatitis B, sífilis, inmune por fármacos, neoplásica.
3. CUADRO CLÍNICO
Sospechar vasculitis en cualquier paciente con síntomas constitucionales (fiebre, astenia, adinamia, fatiga, pérdida de peso, palidez) que tenga evidencia de enfermedad inflamatoria multisistémica. Las manifestaciones clínicas pueden sugerir el tamaño del vaso involucrado y la vasculitis más probable.
Síntomas comunes a todas las vasculitis: fiebre, pérdida de peso, malestar general, artralgias y artritis.
De vasos grandes: claudicación intermitente de miembros, asimetría de la presión arterial, ausencia de pulsos, soplos, dilatación aórtica.
De vasos medianos: nódulos cutáneos, úlceras, lívedo reticularis, gangrena digital, microaneurismas, mononeuritis múltiple.
De vasos pequeños: púrpura, lesiones vasculares ampollosas, urticaria, glomerulonefritis, hemorragia alveolar, granulomas necrotizantes extravasculares cutáneos, hemorragias en astilla, uveítis, epiescleritis, escleritis, SX de ojo rojo.
 |
| Livedo reticularis |
 |
| Hemorragia en astillas |
Las manifestaciones cutáneas son muy importantes y de suma importancia para orientar el diagnóstico: las más habituales son las lesiones purpúricas, que tienen la característica de ser sobreelevadas (púrpura palpable) con o sin centro necrótico. No son pruriginosas ni dolorosas.
Pueden observarse nódulos cutáneos, que a veces siguen los trayectos arteriales; la presencia de livedo reticularis en los miembros inferiores (son más frecuentes ahí por causas hemodinámicas) debe hacernos sospechar de una vasculitis con afectación sistémica siendo frecuente la debilidad, pérdida de peso, hiporexia y decaimiento.
Cualquier cuadro de vasculitis con afectación cutánea puede acompañarse de fiebre, astenia, y/o artromialgias. Se suele referir fiebre leve o adoptar patrones de hipertermia elevada y remitente con escalofríos (fiebre héctica) simulando un cuadro séptico.
Puede presentarse fiebre de origen desconocido, más frecuente en la panarteritis nodosa y arteritis temporal. La aparición de signos y síntomas de insuficiencia cardíaca, arritmias o isquemia miocárdica pueden indicar compromiso vasculítico cardíaco, que suele tener mal pronóstico.
La evaluación inicial de los pacientes en los que se sospecha una vasculitis debe incluir una historia clínica detallada sobre exposición a fármacos y factroes de riesgo para infección por VHB, VHC y VIH. Además la identificación de alguna enfermedad reumática subyacente (artritis reumatoide, LES, etc).
TIP INTERNEURONA:
Arteritis de células gigantes: cefalea o pérdida visual en los adultos mayores; claudicación de la mandíbula (cansancio al masticar); palpación de la arteria temporal (+/-) dolorosa, su pulso puede estar disminuido.
Arteritis de Takayasu: pulsos asimétricos con soplos en un paciente <30 años de edad; pulso arterial ausente; dificultad para medir presión arterial.
Poliarteritis nodosa: mononeuritis múltiple, hipertensión arterial persistente. Dolor testicular.
Vasculitis por ANCA: síndrome pulmonar-renal progresiva (glomerulonefritis/hematuria hemorragia alveolar o hemoptisis). Compromiso de los senos paranasales (sinusitis persistente), otitis media, ojo rojo por escleritis. Churg-Strauss tiene relación con la tríada atópica (rinitis alérgica, dermatitis atópica, asma).
Vasculitis por inmunocomplejos: púrpura palpable.
4. DIAGNÓSTICO
a) Sospecha de SX vasculítico: enfermedad multisistémica, SRIS de causa no definida, fiebre de origen desconocido, afección simultánea o sucesiva pulmonar y renal, tracto respiratorio superior e inferior, manifestaciones isquémicas en jóvenes, síndrome reumatológico con fiebre y compromiso visceral.
b) Evaluar gravedad y extensión del compromiso orgánico: factores pronósticos; tienen peor pronóstico PX con compromiso cardíaco, renal, SNC o intestinal; tienen un curso más benigno si es localizada o lesiones exclusivamente cutáneas.
c) Sospechar imitadores de vasculitis cuando hay: nuevo soplo cardíaco (endocarditis bacteriana subaguda, SBE); necrosis de los dedos de extremidades inferiores (embolias de colesterol); hemorragias en astilla (SBE); disfunción hepática prominente (hepatitis C); abuso de drogas (VIH, hepatitis B/C, cocaína), fiebre inusualmente alta (SBE), historia de actividad sexual de alto riesgo (VIH), DX previo de enfermedad neoplásica.
d) DX enfermedad manifestada por vasculitis, y si es posible, la etiología: determinar si es primaria o secundaria; manifestaciones clínica, calibre de vasos afectados, alteraciones de laboratorio, imagenología e histología.
5. EXÁMENES COMPLEMENTARIOS
Biometría hemática completa: leucocitosis y neutrofilia como expresión del SRIS (PA Nodosa, PA Microscópica, GPA y vasculitis IgA); eosinofilia importante (Churg-Strauss); VSG >100mm y PCR: constante en todos los SX vasculíticos, es infrecuente encontrarla normal; aumento de urea y creatinina (azoados), observar sedimento urinario: compromiso glomerular (hematuria con hematíes dismórficos del 5% o más es glomerular, cilindros hemáticos, leucocituria y proteinuria, que puede ser de rango nefrótico).
Biopsia: la sospecha DX debe corroborarse con el examen histopatológico de los tejidos u órganos comprometidos, como biopsia de lesiones cutáneas, músculo, nervio periférico, pulmón, riñón, etcétera. Esto puede ser granulomatosa, necrosante, o leucocitoclástico con polvillo nuclear o restos fragmentarios leucocitarios.
ANCA: importancia capital en el DX y seguimiento. MPO3-ANCA (+), y eosinófilos o necrosis histológica) Churg-Strauss, si los ANCA son (-) pienso en poliangitis microscópica. PR3-ANCA pienso en granulomatosis de Wegener.
Factor reumatoideo y anticuerpos antinucleares: suelen ser negativos o positivos en título bajo, salvo vasculitis secundaria a AR o LES.
Depósitos de IgA en la prueba de inmunofluorescencia: Vasculitis de Schonlein-Henoch.
Crioglobulinas: crioglobulinemia; investigar presencia de VHC con serología y antigenemia. PAN puede estar asociada con VHB y puede también con distintos tipos de vasculitis el VIH. Pensar en vasculitis crioglobulinémica, muchas veces asociada a hepatitis.
RX o TAC de tórax: infiltrados alveolares focales y nódulos únicos o múltiples, a menudo cavitados. Puede haber derrame pleural o compromiso intersticio-alveolar difuso, sobre todo en casos de hemorragia alveolar (PA Microscópica, crioglobulinémica, vasculitis IgA).
Imagenología vascular: útiles en las vasculitis de grandes vasos y vasos medianos (EcoDoppler dúplex, angiografía digital y angiorresonancia). Podemos encontrar estenosis segmentaria (arteritis Takayasu y de células Gigantes), formación de microaneurismas (mesentéricos, renales) típicos (poliarteritis nodosa), "signo del halo" al rededor de la arteria afectada en el ecoDoppler (arteritis de células Gigantes).
6. TRATAMIENTO
a) Primero, identificar y eliminar agentes incitantes.
b) Tratar la enfermedad subyacente primaria asociada a vasculitis.
c) Iniciar terapia antiinflamatoria y/o inmunosupresora acorde con la extensión de las vasculitis.
C.1) la vasculitis de vasos pequeños confinada a piel, generalmente requiere un tratamiento menos agresivo.
C.2) Primera opción para terapia de inducción de vasculitis generalizada y grave es la combinación de ciclofosfamida y prednisona. Corticoesteroides a dosis altas generalmente como metilprednisolona si hay mucho compromiso de órganos vitales, si no hay compromiso y tiene signos vitales estables se puede dar prednisona y ciclofosfamida.
C.2.1) Agentes biológicos dirigidos (rituximab para vasculitis por ANCA) pueden reemplazar la ciclofosfamida como terapia de inducción.
d) Terapia de mantenimiento con azatioprina, metotrexato o micofenolato mofetilo.
e) Prevenir complicaciones: infecciones (inmunizaciones; profilaxis con TMP/SMX si se administra prednisona a dosis altas), osteoporosis, aterosclerosis (control de la presión arterial y lípidos).
7. VASCULITIS DE GRANDES VASOS
7.1 ARTERITIS DE TAKAYASU
Afecta vasos medianos y grandes (aorta y ramas del arco aórtico). Caracterizado por estenosis, oclusión y, a veces, formación de aneurismas de grandes arterias. Afecta mujeres y hombres por igual, en la 2da o 3ra década de vida.
TIP INTERNEURONA: mayor prevalencia en adolescentes femeninas y mujeres jóvenes asiática; fuerte predilección por el cayado aórtico y sus ramas.
Cuadro clínico: claudicación vascular de nueva aparición en cualquier extremidad (Ej. salir a correr y fatigarse pronto) con anomalías del pulso periférico en un individuo <40 años de edad.
Harrison 20a edición (2019): Síntomas generales, ausencia o disminución de la percepción de pulsos en vasos afectados (frecuente en arteria subclavia), hipertensión.
 |
| Harrison 20a edición (2019) |
Diagnóstico: La biopsia no es la más precisa. Hacer arteriografía aórtica o angiorresonancia, siendo la última potencialmente útil en DX precoz de engrosamiento y edema de la pared aórtica, antes de una estenosis vascular o formación de aneurismas. Criterios de ACR, presencia de 3 o más criterios.
 |
| Criterios de la ACR para Takayasu |
Tratamiento: Corticoesteroides. 40-60mg de prednisona al día > arterioplastía.
Complicaciones: insuficiencia cardíaca congestiva, EVC, IAM, rotura de aneurisma, insuficiencia renal.
TIP INTERNEURONA: Mujer asiática joven con pulsos disminuidos (más a menudo en brazos que en piernas) pienso en Takayasu.
7.2 ARTERITIS DE CÉLULAS GIGANTES
Afecta vasos medianos-grandes de cabeza y cuello. Vasculitis sistémica más común en la edad avanzada. De manera característica abarca una o más ramas de la arteria carótida, en particular la rama temporal. Guarda íntimo vínculo con la polimialgia reumática. Afecta a >50 años, frecuente en mujeres y poco común en raza negra.
Cuadro clínico: cefalea por afectación de arterias intracraneales (76%), pérdida de peso (43%), fiebre (42%), fatiga (39%), cualquier síntoma visual (37%), claudicación de mandíbula (30%, el más específico); puede manifestarse como dolor en la cara o garganta, particularmente al masticar. Asociado comúnmente a polimialgia reumática.
Harrison 20a edición (2019): dolor al tacto, engrosamiento o nódulos; dolor de la piel cabelluda y claudicación del músculo masetero y de la lengua; neuropatía óptica isquémica; apoplejías, estenosis de la arteria subclavia, aneurisma aórtico + manifestaciones de inflamación sistémica (malestar general, fatiga, anorexia, pérdida de peso, hiperhidrosis, artralgias, polimialgia reumática o afectación de grandes vasos.
Complicación más común: amaurosis fugaz.
Diagnóstico: laboratorios con VSG y PCR elevados; biopsia de arteria temporal. Tres o más criterios de la clasificación de la ACR. Podemos encontrar anemia normocrómica o levemente hipocrómica y aumento de la fosfatasa alcalina.
 |
| Criterios de la ACR de Células Gigantes |
Tratamiento: Corticoesteroides iniciados en el momento de sospecha del diagnóstico sin esperar resultados de la biopsia, inicialmente 1mg/kg/día. También con metotrexato, calcio, vitamina D y bifosfonatos.
Esquema Harrison 20a edición (2019): Prednisona 40-60mg/día durante un mes para luego reducir, durante 2 años o más. En caso de afección visual, considerar 1000mg de metilprednisolona durante tres días. Prednisona 10-20mg/día en caso de polimialgia reumática. AAS 81mg disminuye complicaciones; tocilizumab (antirreceptor de IL-6), Abatacept (CTLA4-Ig).
Pronóstico: Recaídas del 25-50% al disminuir la medicación en los pacientes.
8. VASCULITIS DE VASOS MEDIANOS
8.1 POLIARTERITIS NODOSA
Arterias musculares de tamaño mediano afectadas, venas preservadas. Puede dañar cualquier órgano del cuerpo. Es más común en hombres 2:1, y en personas de la 5ta-6ta década de la vida. No abarca arterias pulmonares, aunque puede abarcar bronquiales. No se observan granulomas, eosinofilia significativa o diátesis alérgica.
Cuadro clínico: inicio subagudo de enfermedad inflamatoria multisistémica, úlceras en piernas, orquitis, dolor abdominal (abdominalgia por isquemia abdominal), hipertensión sin evidencia de afectación glomerular o alveolar. Debilidad (69%), pérdida de peso (67%), mononeuritis múltiple (42%), polineuropatía (35%), azoemia (40%), hipertensión arterial sistólica (37%). Tres o más criterios de la clasificación de la ACR. Pueden haber púrpura palpable, microaneurismas; no afecta pulmón ni causa glomerulonefritis (lesión renal glomerular) pero sí aumenta creatinina.
 |
| Criterios de la ACR poliarteritis nodosa. |
 |
| Harrison 20a edición (2019) |
Histopatología: necrosis e inflamación intercaladas con vasos sin afección.
Diagnóstico: biopsia de órganos afectados, si no son de fácil acceso es útil el arteriograma con medio de contraste dirigido con catéter.
Laboratorios: PCR elevada, hipergammaglobulinemia, ANCA (-), leucocitosis con predominio de neutrófilos en un 75% de PX, anemia normocrómica, aumento de VSG. Buscar hepatitis B y C.
Tratamiento: prednisona (pilar), metilprednisolona (exacerbaciones), ciclofosfamida, azatioprina. Esquema de prednisona + ciclofosfamida.
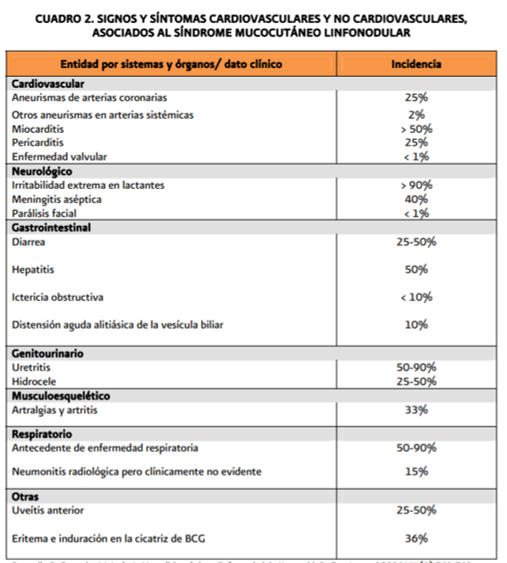
8.2 ENFERMEDAD DE KAWASAKI
También llamado síndrome mucocutáneo linfonodular. Entidad aguda, febril, que afecta múltiples órganos en niños. En los casos graves se encuentran vasculitis de arterias coronarias. Se identifican aneurismas y trombosis a manera de cuentas. 80% de los casos se dan antes de los 5 años de edad, y la incidencia máxima en <2 años. Se asocia a arteritis de los vasos grandes o medianos.
Patogenia: proliferación e infiltración de la íntima de la pared vascular por mononucleares.
Cuadro clínico: adenitis cervical no supurada bilateral, exantema polimorfo, congestión de conjuntivas, eritema de cavidad bucal/labios/palmas, descamación de la piel de la yema de los dedos, aneurismas coronarios y aórticos así como trombosis.
 |
| Manifestaciones Clínicas |
 |
| Criterios de la AHA 2017 |
 |
| Manifestaciones de Kawasaki |
 |
| Criterios diagnósticos de Kawasaki |
Tratamiento: gammaglobulina IV 2g/kg en 10h, AAS 100mg/kg/día por 14 días, seguido de 3-5 mg/kg al día durante varias semanas. Alternativa: cirugía.
9. VASCULITIS DE VASOS PEQUEÑOS
9.1 MEDIADO POR ANCA
Los ANCA's son anticuerpos dirigidos contra proteínas específicas en gránulos en el citoplasma de neutrófilos y proteínas lisosomales en monocitos, y están presentes en los sueros de pacientes. Hay tres tipos importantes: proteinasa-3 (PR3), mieloperoxidasa (MPO), proteína 2 de membrana asociada a lisosoma (LAMP2).
9.1.1 GRANULOMATOSIS CON POLIANGITIS (SX DE WEGENER)
Tiene 3 características principales: compromiso del tracto respiratorio superior e inferior (sinusitis crónica) con vasculitis granulomatosa de los vasos pequeños y necrosis; glomerulonefritis focal/segmentaria necrotizante; asociación con c-ANCA y anti-PR3. Tiene un pico o edad media de presentación a los 41 años.
Puede ser localizada (10%) con afección respiratoria alta + baja + riñones + ANCA (+) en un 90% de PX o localizada sin compromiso renal + ANCA (+) en un 60% de PX.
Cuadro clínico: Senos paranasales (sinusitis crónica 80% portadores de S. aureus); Mucosa nasal (inflamación crónica purulenta 70%, epistaxis, ulceraciones, perforación e interrupción del tabique/cartílago nasal); Mucosa oral (inflamación crónica con úlceras orales que pueden ser, o no, dolorosas); Mucosa Faríngea (inflamación crónica que obstruye trompa de Eustaquio y provoca OM supurativa o serosa crónica); Mucosa laríngea y traqueal (Ronquera, estenosis subglótica con estridor, insuficiencia respiratoria); Entre otras como ulceraciones en la mucosa bucal o nasal, pseudotumores, conjuntivitis, epiescleritis, destrucción del cartílago nasal (en silla de montar, igual que en sífilis), epistaxis, infiltrados pulmonares nodulares que pueden cavitarse (NO todos los ocupamientos pulmonares son neumonía infecciosa), hemorragia alveolar, púrpura, glomerulonefritis, pericarditis.
Afección pulmonar (90%): Infiltrados cavitados nodulares bilaterales (vasculitis granulomatosa necrosante), granulomas (causan insuficiencia respiratoria; hay nódulos fijos a comparación de Chugh-Strauss), capilaritis (causan hemorragia alveolar por infiltración neutrofílica y necrosis fibrinoide de septos alveolares), fibrosis (por resolución de la inflamación crónica).
Manifestaciones renales (50-80%): glomerulonefritis necrozante pauci-inmune, focal, segmentaria, puede haber o no granulomas, no hay inmunocomplejos. Sedimento activo con hematuria (eritrocitos dismórficos), piuria, proteinuria, cilindros celulares. Todo esto más alteración de la función renal evaluada con creatinina.
Manifestaciones oculares (30-60%): proptosis por infiltración fibrótica del espacio retro-orbital (lesión del nervio óptico), escleritis, epiescleritis, uveítis.
Manifestaciones en músculo esquelético (67%): artralgias, mialgias, sinovitis sin destrucción articular.
Manifestaciones cutáneas (40-50%): púrpura, úlceras, nódulos subcutáneos, vesículas. Histología con vasculitis necrotizante con o sin infiltración granulomatosa.
SNC y SNP (15-10%): es característico la mononeuritis múltiple, polineuropatía simétrica. La mononeuritis múltiple es una neuropatía que involucra nervios sensitivos y motores aislados, normalmente de inicio agudo; afecta con frecuencia e intensidad a los nervios finbulares; puede progresar a una polineuropatía; afecta principalmente al orden motor sobre sensitivo tanto miembros superiores como inferiores.
Manifestación cardíaca (5%) frecuente es la pericarditis.
 |
| Manifestaciones del SX de Wegener |
 |
| Harrison 20a edición (2019) |
Los pacientes pueden presentar infecciones por agentes oportunistas (P. jiroveci, S. pneumoniae, herpesvirus, legionella, entre otros).
 |
| Criterios de la ACR para SX de Wegener |
Laboratorios: C-ANCA / Anti-PR3 > Anti MPO.
Tratamiento: terapia de inducción (peligro para la vida, compromiso de órganos, enfermedad que no amenaza los órganos) con prednisona o metilprednisona + terapia de remisión 6 meses después con ciclofosfamida.
Esquema: metilprednisolona IV 15mg/kg/día por 3-5 días más ciclofosfamida IV pulso mensual 0.5-1g/m2. Al usar prednisona, reducir gradualmente (no <15mg/día en el primer mes). El metotrexato VO 0.3mg/kg semanal, aumentar la dosis 2.5mg hasta llegar a 20mg y mantener. Azatioprina 2mg/kg/día.
Esquema Harrison 20a edición (2019):
a) Inducción
- Amenaza la vida: metilprednisolona por 3-5 días + plasmaféresis + ciclofosfamida.
- Amenaza órganos: metilprednisolona + Ciclofosfamida IV // Rituximab eliminada por plasmaféresis.
- Sin amenaza de órgano: prednisona 1mg/kg/día (disminución a 15mg/día en el primer mes) + metotrexate.
- Si hay vasculitis generalizada: ciclofosfamida / rituximab.
b) Remisión
- Cambio a otros inmunosupresores a los 6 meses: azatioprina, metotrexate, micofenolato, leflunomida.
- Terapia agresiva: gammaglobulina IV, infiximab con o sin trasplante renal.
- Mantener en TX independiente de la remisión clínica y analítica.
c) Profiláctica
- TMP/SMX por jirovecii.
- Bactroban 2% intranasal.
- Profilaxis para osteoporosis por corticoesteroides.
 |
| TX según la GPC |
9.1.2 POLIANGITIS MICROSCÓPICA
Vasculitis necrotizante sistémica asociada a glomerulonefritis necrotizante segmentaria focal y capilaritis pulmonar. No causa formación de granulomas o vasculitis granulomatosa. Afecta a ambos sexos y personas de 30-50 años.
TIP INTERNEURONA: paciente con síndrome pulmonar-renal, no granulomatosa, asociado a P-ANCA y MPO-ANC en un 60%.
Cuadro clínico: capilaritis pulmonar (hemoptisis) y glomerulonefritis necrotizante segmentaria y pauci-inmune (hematuria), con o sin P-ANCA (+). Biopsia pulmonar con capilaritis funcional pero con inmunofluorescencia negativa.
Diagnóstico: Cuadro clínico característico + Biopsia renal con glomerulonefritis necrotizante sin depósitos inmunes + P-ANCA contra MPO (apoya DX).
Tratamiento y pronóstico: Signos clínicos similares a GPA y tratamiento similar al usado en VGPA; sobrevida a 5 años es de 74% y la mortalidad es frecuente por hemorragia alveolar.
Laboratorios: P-ANCA / Anti MPO > Anti-PR3
9.1.3 GRANULOMATOSIS EOSINOFÍLICA CON POLIANGITIS (CHURG-STRAUSS)
Vasculitis de vasos pequeños que afectan piel, nervios periféricos y pulmones. Hay reacciones granulomatosas en los tejidos y pared de los vasos. Incidencia de 3 por cada millón de habitantes. La edad media de comienzo es de 48 años. Es común la eosinofilia periférica; antecedentes de rinitis alérgica en un 70% y de asma en un 95%.
TIP INTERNEURONA: tríada clásica de asma intensa, eosinofilia y vasculitis.
Cuadro clínico: lesiones cutáneas en forma de nódulos cutáneos y subcutáneos llamados granulomas extravasculares de Churg-Strauss, caracterizados por áreas de necrosis del colágeno rodeados de infiltrado granulomatoso. El síntoma inicial hasta en un 85% de los PX es la disnea. Al igual que todas las vasculitis, síndrome constitucional de fiebre, astenia, adinamia, pérdida de peso. Pero en general consta de tres fases; fase prodrómica (28 meses a 7años) con manifestaciones alérgicas de rinitis poliposis y asma progresiva, fiebre en 50%; fase de desarrollo: eosinofilia periférica, neumonía eosinofílica crónica o gastroenteritis eosinofílica, miocarditis; fase vasculo-sistémica: asma disminuye abruptamente, desarrollo de miocarditis, insuficiencia valvular, neuropatía periférica vasculítica, gastroenteritis eosinofílica, púrpura y dolor testicular.
TIP INTERNEURONA: afección pulmón > nervios periféricos > piel > miocardio.
 |
| Manifestaciones clínicas |
Laboratorios: P-ANCA / Anti MPO > Anti-PR3; eosinofilia >1500 células/microlitro, anemia por enfermedad crónica, VSG y PCR aumentadas, IgE (70%), factor reumatoide positivo(70%), ANCA´s (50-65%) con mayor probabilidad de desarrollar enfermedad renal, hemorragia alveolar, mononeuritis múltiple y púrpura.
Diagnóstico: cuadro clínico + eosinofilia >1500células/microlitro + vasculitis sistémica que involucre 2 o más órganos + confirmación por biopsia del tejido afectado con granulomas necrotizantes extravasculares cerca de arterias pequeñas y vénulas con macrófagos y células gigantes.
Tratamiento y pronóstico: PX con cuadro sin afectación fulminante multiorgánica dar esquema con glucocorticoides (prednisona) disminución gradual a menos que lo necesite por el asma; PX con afección multiorgánica dar combinación de prednisona con ciclofosfamida. Con tratamiento hay sobrevida a 78 meses en el 72% y sin tratamiento la sobrevida a 5 años es del 25%. Mepolizumab (anti IL-5) puede funcionar para asma recurrente.
TIP INTERNEURONA: Churg-Strauss tiene infiltrados pulmonares migratorios, antecedente de alergias, rinitis, asma , eosinofilia, P-ANCA y es de buen pronóstico VS Wegener con infiltrados pulmonares cavitados, epistaxis, nariz en silla de montar, estenosis traqueal, glomerulonefritis rápidamente progresiva, C-ANCA y es de mal pronóstico.
9.2 MEDIADO POR INMUNOCOMPLEJOS
9.2.1 CRIOGLOBULINÉMICA
Mediada por crioglobulinas, caracterizada por depósito de inmunocomplejos, dependiente de la temperatura y subsecuente inflamación de la pared de vasos, infección por VHC en un 70-90%.
TIP INTERNEURONA: el dato fundamental es la presencia de crioprecitipitados circulantes; en el 90% hay hipocomplementonemia; importante buscar VHC.
Cuadro clínico: vasculitis cutánea (púrpura palpable, ulceración, necrosis), síntomas vasomotores (fenómeno de Raynaud, acrocianosis), artritis, neuropatía periférica, glomerulonefritis.
Diagnóstico: presentación clínica + elevación de crioglobulinas + factor reumatoide (+)/ANA (+)/Hipocomplementonemia de C3 y C4.
Tratamiento: antivirales contra hepatitis C en su caso, glucocorticoides y ciclofosfamida (o rituximab) para casos graves; plasmaféresis en enfermedad severa por complicaciones graves o potencialmente mortales por crioprecipitación o hiperviscosidad sérica con uso concomitante de corticoesteroides a dosis altas. En crioglobulinemia asintomática no requiere tratamiento. Los AINES se pueden usar en PX con artralgia y fatiga. El IFN-alfa pegilado con ribavarina ha demostrado eficacia en pacientes con crioglobulinemia asociada a VHC.
9.2.2 PÚRPURA DE SCHÖNLEIN-HENOCH
Depósito de inmunocomplejos en vasos finos. Cuadro de curso agudo y resolución rápida (97%, en 6-16 semanas) de púrpura palpable, artralgias, signos y síntomas digestivos y glomerulonefritis. El pico es a los 2-10 años en un 90%, pero se suele observar en niños de 4-7 años. En los adultos es común la afección renal.
Etiología: nuevos medicamentos, infecciones respiratorias recientes, FR para VHC, 30-50% sin causa identificable.
Cuadro clínico: púrpura palpable, poliartritis sin artritis franca, afectación del tubo digestivo en un 70% (dolor abdominal tipo cólico, náusea, vómito, diarrea, estreñimiento, expulsión de sangre y moco por el recto). La afectación al miocardio se observa en adultos, pero es rara en niños.
Diagnóstico clínico y tratamiento de glucocorticoides en casos severos, pero por lo general es autolimitado.
TIP INTERNEURONA: tétrada de púrpura palpable, artritis (70%), dolor abdominal (70%) y afección renal (50%).
9.2.3 OTROS Y SECUNDARIOS
Se encuentra la urticaria vasculítica, hipocomplementonemia, vasculítis lúpica, vasculitis reumatoide, colitis ulcerosa, sarcoidosis.
Secundarios fármaco-inducido por alopurinol, tiazidas, sulfonamidas.
Secundarios por infección en la enfermedad del suero, rickettsiosis, VEB.
10. ANEXOS
 |
| Harrison 20a edición (2019) |
 |
| Harrison 20a Edición (2019) |


11. ¿CÓMO CITO ESTE ARTÍCULO PARA MIS TRABAJOS?
Medina I., Todo sobre vasculitis: Repaso ENARM [Internet]. México: Interneuronamed; 2021 jul. [Consultado - Revisado]. Disponible en: https://interneuronamed.blogspot.com/2021/10/Vasculitis.html
Modificar la sección de "[Consultado - Revisado]" con la fecha de hoy como alguno de los ejemplos siguientes:
1. [Revisado 01 jul 2021]
2. [Consultado 01 jul 2021]
