

LINFOMAS HODGKIN Y NO HODGKIN

ÍNDICE
1. GENERALIDADES Y CONCEPTOS BÁSICOS
¡Bienvenidxs!, soy Interneuronamed y te dejaré los puntos clave para comprender los linfomas.
Sígueme en mis redes sociales (Twitter, Facebook, YouTube, Instagram) como @Interneuronamed, donde encontrarás resúmenes de medicina y más cosas que interesarán.
1. GENERALIDADES Y CONCEPTOS BÁSICOS
Un linfoma es un cáncer del sistema linfático, el cual ataca en particular a los linfocitos. Se puede dividir en Hodgkin (HL) y No Hodgkin (NHL).
2. LINFOMAS HODGKIN
Linfoma Hodgkin (18%): Es una neoplasia hematológica de los linfocitos B descrita por primera vez por el patólogo Thomas Hodgkin en el siglo XIX. En México es una neoplasia poco frecuente en adultos (1%), más frecuente en niños y es más frecuente en varones. Tipo de linfoma más frecuente entre los 15-19 años (infancia y 2da década de la vida). Son clasificados según la morfología e inmunohistoquímica, con presentación bimodal a los 15-34 años y >55 años. Curable en >80% y recaídas en 10-30%.
TIP INTERNEURONA: se caracteriza por presentar células de Reed-Sternberg a comparación de su ausencia en el No Hodgkin.
 |
| Infografía del día mundial de Linfoma Hodgkin |
- Factores de riesgo HL: adulto joven o adulto mayor; sexo masculino; antecedente de infección por VEB; familiar de primer grado con HL; mononucleosis infecciosa; tabaquismo; VIH; inmunosupresión; trasplante de órgano sólido; enfermedades autoinmunes; TAC en la infancia o adolescencia; factores ambientales en trabajadores de madera, agricultores y procesadores de carne.
Se ha detectado ácido nucleico de VEB hasta en un 20-50% de los casos asociado a celularidad mixta (75%) y depleción de linfocitos (75%). Los hermanos del mismo sexo de pacientes con LH tienen 10 veces más probabilidades de tener LH.
- Factores protectores: varicela, sarampión, rubéola, tos ferina, paperas, aspirina.
TIP INTERNEURONA: riesgo absoluto de desarrollar LH con VEB (+) después de mononucleosis infecciosa 0,1%. Aumento 4 veces el riesgo de HL positivo para EBV.
- La OMS lo clasifica en HL clásico (95%) con cuatro subtipos: esclerosis nodular (70%), celularidad mixta (20-25%), predominio linfocitario (5%) y depleción linfocitaria (<1%); y HL con predominio linfocitario nodular (5%, célula en palomita de maíz). En el tercer mundo, la celularidad mixta es más frecuente en niños y adultos.
- Cuadro clínico: adenopatías no doloras supra-diafragmáticas (clavicular y supraclavicular más frecuentes), prurito generalizado e intenso (mal pronóstico), signo de Hoster y tríada o síntomas B (20% en estadio I/II, y 50% en estadio III/IV): fiebre, sudoración nocturna y pérdida de peso >10% en 6 meses. Pueden presentarse con síndrome de vena cava (ingurgitación y edema de la cara y tórax superior), derrame pericárdico / pleurales.
- Signo de Hoster: dolor de adenopatías o ganglios linfáticos con la ingesta de bebidas alcohólicas. Es más frecuente en la esclerosis nodular.
- Síndrome de Bazex: acroqueratosis paraneoplásica.
 |
| Acroqueratosis paraneoplásica |
- Otros síntomas y síndromes asociados: enfermedad intra-abdominal, colestasis hepática, dolor inducido por alcohol (1-2%), lesiones cutáneas, síndromes neurológicos (0,05%) como corea, encefalitis, SX de motoneurona inferior, síndrome nefrótico, hipercalcemia, anemia, eosinofilia, leucocitosis, trombocitosis, linfopenia.
- Estudios diagnósticos: estudios de imagenología tórax y cuello con radiografías, USG, TAC, u otros; estudios de laboratorio: BH completa (anemia, leucocitosis, linfopenia), QS y VSG elevada; biopsia excisional e inmunohistoquímica (célula de Reed-Sternberg). Otras como fosfatasa alcalina, DHL, perfil de función hepática y albúmina, ácido úrico, VHC, VHB y VIH. La PET-CT se usa comúnmente para evaluar la extensión y determinar la etapificación del linfoma, puede ser positiva en inflamación o infección sin LH; es positiva en médula ósea si hay 3 o más lesiones multifocales y negativa si la captación es homogénea.
TIP INTERNEURONA: la biopsia excisional es el estándar de oro diagnóstico.
- Estudios de extensión: TAC contrastada de cuello, tórax, abdomen y pelvis. PET-CT para estadificación inicial y seguimiento, diferenciar enfermedad residual de fibrosis o necrosis. AMO y biopsia de hueso si no está disponible PET-CT y el PX está en estadio clínico IIB, III o IV.
En radiografía de tórax hay presencia de ensanchamiento de mediastino con múltiples imágenes nodulares en ambos campos pulmonares. La tráquea con disminución de calibre de la luz y borramiento del contorno cardíaco.
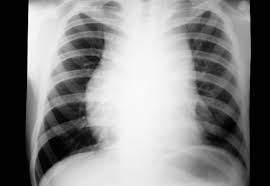 |
| Radiografía de Linfoma Hodgking |
Ejemplo de un caso: Hb 8.5 g/dL, hematocrito 28%, leucocitos 21,000 células/mm3, neutrófilos segmentados 55%, eosinófilos 8%, basófilos 1%, linfocitos 30%, monocitos 6%, VSG 96 mm/1h, albúmina 4g/dL, DHL 1469 UI/L.
La célula de Reed-Sternberg pierden las características fenotípicas de las células B del centro germinal, no expresan BCL6 y no muestran evidencia de hipermutación somática o transcripción del gen de Ig. Son células grandes con citoplasma abundante, levemente basófilo, núcleos bilobulados, dobles o múltiples e inclusiones eosinofílicas en nucléolos.
Eliminan su programación de expresión génica de células B por metilación del ADN, regulación positiva de NOTCH1 y del alza de proteínas de linaje no-B como ID2, ABF1. La vía del INF-kB con inflamación y microambiente tumoral que promueven la supervivencia de las células de Reed-Sternberg del ataque de los linfocitos T.
-Patogénesis: mecanismo de estimulación por producción de quimiocina CC17 que atrae LT liberando TNF-a y CD40+, y mecanismo de escape por citoquinas inmunosupresoras PDL-1 y PDL-2 con pérdida de la expresión de HLA que evita la activación de LT.
TIP INTERNEURONA: el aspirado por aguja fina no es útil en esta enfermedad.
 |
| Célula de Reed-Sternberg |

Para estadificar a los pacientes se utiliza la clasificación de Ann Arbor:
 |
| Clasificación de Ann Arbor |


- Tratamiento: quimioterapia con esquema ABVD (doxorrubicina, bleomicina, vinblastina y dacarbazina) + radioterapia. En casos de recidivas se maneja con trasplante de células madre.
En estadio intermedio, cuatro ciclos de ABVD seguido de radioterapia fraccionada convencional a 30Gy es el estándar de oro.
En estadio alto el tratamiento sólo con quimioterapia, la radioterapia es para pacientes con enfermedad residual después de la quimioterapia.
TIP INTERNEURONA: Evaluar criterios para quimioterapia y complicaciones del TX. Si tiene síntomas B, dar quimioterapia. Hay nuevos esquemas como el BEACOPP, COPP/ABVD, MOPP, STANFORD V. Estudios complementarios son pruebas de función pulmonar, ecocardiograma, asesoramiento reproductivo, acumulación de esperma y recolección de ovocitos.
- Complicaciones por radioterapia: (tempranas) sequedad de la boca, faringitis, tos, dermatitis, anorexia, náuseas, mielosupresión y trombocitopenia; (tardías) hipotiroidismo, pericarditis, neumonitis, signo de Lhermitte, arteriopatía coronaria, segundas neoplasias en pulmón, mama, tiroides y estómago.
- Complicaciones por quimioterapia: (tempranas) náuseas, vómito, alopecia, mielodepresión, infección; (tardías) esterilidad (MOPP), neuropatía (vincristina), miocardiopatía (doxorrubicina), fibrosis pulmonar (bleomicina), leucemia secundaria (BEACOPP,MOPP + RT).
- Pronóstico: cuando hay factores favorables la supervivencia es del 87%, disminuye a 60-70% cuando hay factores adversos (edad, mal estado de salud, Hb <10.5g/dL, estadio IV, leucocitos >15,000/mm3). Con el tratamiento moderno hasta el 80-90% de los pacientes logran remisión permanente. Será desfavorable (I/II) en VSG >50 s/ síntomas B, >50 años, >3 focos de afección ganglionar, >1 área de afectación extraganglionar y presencia de síntomas B.
- Puntuación pronóstica (1 punto por cada factor positivo): Hb <10.5g/dL, sexo masculino, edad mayor igual a 45 años, estadio IV, leucocitos >15,000/mm3 o recuento absoluto <600/mm3 o <8% del total, albúmina <4g/dL. Entonces la supervivencia a 5 años será: 84% con ningún factor, 77% con un factor, 67% con dos factores, 60% con tres factores, 51% con cuatro factores y 42% con más de cinco factores.
- Seguimiento: detectar recurrencias (cada 3 meses en el primer año luego cada 6 meses por 4 años después de un año de tratamiento), evaluar función tiroidea, niveles de estrógenos y testosterona por la radiación.
a) ESCLEROSIS NODULAR
Es el subtipo más común y segundo subtipo con mejor pronóstico. Incide en adolescentes y adultos jóvenes causando afección mediastinal y supra-diafragmática (más frecuentes), patrón parcialmente nodular por presencia de bandas fibrosas junto a áreas difusas. La célula característica es variante lacunar de Reed-Sternberg.
b) CELULARIDAD MIXTA
Es el subtipo con tercer mejor pronóstico, más frecuente en niños y adultos mayores. Prevalece en pacientes con VIH. Consta de infiltrado de fondo con eosinófilos, neutrófilos, histiocitos y células plasmáticas. Se caracteriza por crecimiento difuso nodular sin bandas de fibrosis y tiene células de Reed-Sternberg.
c) LH RICO EN LINFOCITOS
Es el subtipo con mejor pronóstico, es frecuente detectarlo en etapa temprana. Presenta células de R-S, células de Hodgkin mononucleares y células que se asemejan a linfocitos histiocíticos. Patológicamente hay infiltrado de predominio linfocitario con eosinófilos o neutrófilos raros o nulos, crecimiento nodular difuso. Debe hacerse diferencial con LHPLN.
d) LH DEPLECIÓN LINFOCITARIA
El subtipo más raro, agresivo y de peor pronóstico. Se presenta en adultos mayores y pacientes con VIH. Es frecuente encontrarlo en etapas avanzadas. Las células de R-S adquieren apariencia sarcomatosa. En patología hay apariencia hipocelular por fibrosis, necrosis y células inflamatorias pauci-inmunes.
e) LH NO CLÁSICO
Se expresan marcadores de células B (CD20+, CD45+, BCL6+). No se relaciona con VEB, el 80% es una enfermedad autolimitada. Presenta linfadenopatía crónica periférica asintomática en el 100% de los casos. Y tiene como característica células en forma de palomitas de maíz.
3. LINFOMAS NO HODGKIN
Neoplasia maligna que afecta al sistema linfático y frecuentemente se extiende a sitios extranodales. Son cánceres de linfocitos B (80-85%), linfocitos T (15-20%) y NK maduros (infrecuente). Su pronóstico y evolución natural tienden a ser más variables.
En México representan la 2da causa de neoplasias hematológicas y el 6to lugar de muerte por cáncer en el mundo. Su incidencia >50% en varones sobre mujeres, suele afectar a cualquier edad, pero sobre todo a partir de los 3 años, sin embargo a mayor edad mayor incidencia. La edad promedio de diagnóstico es de 45-55 años, con incremento gradual del riesgo a partir de los 50 años.
TIP INTERNEURONA: 5 veces más frecuente que el linfoma Hodgkin; los pacientes con inmunodeficiencia primaria y secundaria están predispuestos a linfomas no Hodgkin.
Los linfomas de linfocitos T son más frecuentes en Asia, mientras que ciertas variedades de linfomas de linfocitos B, como el linfoma folicular, aparecen con frecuencia en países occidentales. El linfoma angiocéntrico de linfocitos NK es más frecuente en el suroeste asiático y algunas partes de hispanoamérica.
- Etiología: inmunodeficiencias previas (hereditario, adquirido, enfermedades autoinmunes como LES, QT y RT previas); agentes infecciosos (VEB, VHC, VIH, HTLV-1, H. pylori, C. jejuni; agentes ambientales (productos agroquímicos, fármacos, transfusiones); alteraciones citogenéticas como el linfoma folicular (BCL-2) y linfoma difuso de células B grandes (BCL-6) que son de afección de la zona folicular.
- Factores predisponentes: inmunodeficiencias hereditarias (SX de Klinefelter, SX de ataxia-telangiectasia, entre otras) y adquiridas (inmunodepresión yatrógena, infección por VIH-1, hipogammaglobulinemia adquirida), enfermedades autoinmunitarias (SX de Sjögren, esprúe celíaco, AR, LES), exposición a sustancias químicas o fármacos (difenilhidantionato, dioxina, fenoxiherbicidas, RT Y QT, radiación.
- Inmunología: alrededor del 90% de los linfomas proceden de linfocitos B, el más frecuente es el de células grandes seguido del folicular. Casi todos los linfomas de linfocitos B surgen después de alteraciones del gen de inmunoglobulinas. La fase de diferenciación de un linfoma maligno no predice su evolución natural, sin embargo, es útil para el DX y permite identificar subtipos específicos.
- Clasificación:
Linfomas de bajo grado o poco agresivos (linfoma folicular) que suelen estar diseminados en el momento del DX, son de crecimiento lento y poco sintomáticos. Es difícil que alcancen la remisión completa por baja sensibilidad a QT (quimioterapia). Pueden transformarse a una forma histológica más agresiva.
Linfomas de alto grado o agresivos (linfoma B difuso de células grandes) que es de rápido crecimiento y mucha sintomatología; aparecen metástasis en diversos órganos, el pronóstico es malo pero la remisión completa se produce en el 80% con tratamiento. Uno de los linfomas muy agresivos es el linfoma de Burkitt.
 |
| Clasificación de la OMS |
TIP INTERNEURONA: Estirpe B, Fácil (folicular), Difícil (difuso), Más o menos difícil (manto), Burkitt. Estirpe T, Mi-Ana (Micosis, Anaplásico).
- Cuadro clínico: linfadenopatía indolora, esplenomegalia o masa extranodal. Algunos pacientes estarán asintomáticos con agrandamiento de nódulos linfáticos como hallazgo incidental en la exploración física o radiológica. Un 25% de los PX pueden tener o no síntomas B, hepatoesplenomegalia, manifestaciones extraganglionares en piel, tracto GI, cabeza, cuello y SNC.
Linfomas indolentes: linfadenopatía periférica, asintomática frecuentemente a menos que cause compresión (uréter, órbita, médula espinal).
Linfomas agresivos: rápido crecimiento, sintomáticos, manifestados como única masa en el pecho o abdomen, extensión a ganglios a distancia sin afectación contigua y LDH elevada.
- Diagnóstico: Es histopatológico principalmente. BH, QS, electroforesis de proteínas, PFH (si está alterado, no iniciar QT por hepatotoxicidad), Rx de Tórax (ganglios linfáticos agrandados en tórax y mediastino). Otras pruebas que se pueden realizar: VIH en linfoma de alto grado, endoscopía en linfoma gástrico tipo MALT, muestreo de líquido pleural o peritoneal (toracocentesis y paracentesis), B2-microglobulina en seguimiento de NHL.
- Pruebas para evaluar grado de extensión/diseminación: biopsia excisional del nódulo linfático guiada por USG/TAC, TAC tórax y abdomen, PET-CT, inmunohistoquímica.
Para la estadificación clínica se utiliza la clasificación de Ann-Arbor modificada por Costswolds, buscando los sitios nodales o extranodales afectados por la enfermedad. Hay que evaluar exploración física, confirmación de síntomas B, estudios de laboratorio (BH completa, PFH, ácido úrico, calcio, electroforesis de proteínas séricas, B2-Mi), RX de tórax, CT de abdomen, pelvis y tórax, biopsia de médula ósea, punción lumbar, gammagrafía con galio o PET.
 |
| Clasificación de Ann Arbor |
- Pronóstico: índice pronóstico internacional (IPI), depende del diagnóstico histopatológico. Hay curación del 30% en adultos y 70-90% en niños.
 |
| Harrison 20a edición |
En linfomas de alto grado:
TIP INTERNEURONA: ELENA tiene linfoma (Edad >60 años, LDH elevada, Estadio Ann Arbor III-IV, número mayor igual a 2 áreas extraganglionares, afectación del estado general mayor igual a 2 (ECOG) o menor igual a 70 (Karnofsky).
La sobrevida a 5 años es del 73% con riesgo bajo cuando tienen 0-1 criterios, del 51% con riesgo bajo-intermedio cuando tienen 2 criterios, del 43% con riesgo alto-intermedio cuando tienen 3 criterios y del 26% con riesgo alto cuando tienen 4-5 criterios.
En linfomas de bajo grado:
TIP INTERNEURONA: HELEN tiene linfoma (Hb <12g/dL, edad >60 años, LDH elevada, estadio Ann Arbor III-IV, número de áreas ganglionares mayor igual a 5.
La sobrevida a 10 años es del 71% con riesgo bajo con 0-1 criterios, de 51% con riesgo intermedio con 2 criterios, y del 36% con riesgo alto con 3 o más criterios.
- Tratamiento en linfomas indolentes o de baja agresividad: en PX asintomático y estadios no avanzados, una opción es el TX conservador hasta la aparición de síntomas; en estadios avanzados QT en monoterapia o combinados, TX tradicional de clorambucilo o QT poco agresivas asociadas a esteroides.
- Tratamiento de linfomas agresivos: se basa en ciclos de poliquimioterapia agresiva como CHOP (ciclofosfamida, adriamicina, vincristina y prednisona), R-CHOP (Rituximab + CHOP) en PX con anti-CD20+ MACOP-B (metotrexate, doxorrubicina, ciclofosfamida, vincristina, bleomicina, prednisona, ácido fonílico) asociado a Rituximab o anti CD20 en linfomas B con o sin autotrasplante de progenitores hematopoyéticos.
a) Linfoma de Burkitt
<1% de linfomas no Hodgkin, pero 30% de los linfomas no Hodgkin en niños; proliferación más rápida y tiempo de duplicación <24h. Hay una forma endémica africana de tumor en el maxilar inferior o huesos de la cara que se propaga a sitios extraganglionares (ovarios, testículos, riñones, mamas, médula ósea, meninges).
La forma no endémica tiene cuadro inicial abdominal con enfermedad masiva, ascitis, afectación renal, testicular/ovárica o ambos, y se propaga a médula ósea y SNC como en la forma endémica. El índice de proliferación es cercano a 100% con macrófagos en cielo estrellado, propios del tumor.
b) Linfoma difuso de linfocitos B grandes
La neoplasia consiste en una masa de linfocitos atípicos grandes con elevado índice de proliferación.
Es el subtipo histológico más frecuente de NHL y representa cerca del 33% de los casos, es un conjunto heterogéneo de múltiples entidades. Mediana de edad al momento de diagnóstico de 64-70 años. Es frecuente en varones, raza blanca y 25% en niños.
Cuadro inicial avanzado en su estadio, solo 30-40% se trata en estadio I o II. Hasta 40% de los PX sufren afectación en sitios extraganglionares como médula ósea, SNC, tubo digestivo, tiroides, hígado y piel.
La QT combinada permite una curación cualquiera que sea el estadio de la enfermedad. La adición del Rituximab (Ab-CD20+) al régimen de R-CHOP mejora la supervivencia. Tiene el peor pronóstico.
c) Linfoma folicular
Los linfomas foliculares ocupan el segundo lugar en frecuencia entre las entidades NHL, en EE.UU. y Europa comprenden hasta el 22% de este tipo de neoplasias en el mundo. Una biopsia adecuada analizada por hematólogo experto es suficiente para establecer DX de linfoma folicular. Es muy sensible a RT y QT. La edad media del diagnóstico es a los 59 años.
El tumor está formado por un porcentaje variable de linfocitos pequeños hendidos y linfocitos grandes, cuyo crecimiento sigue un patrón de aspecto folicular. La manifestación más frecuente son adenopatías indoloras de aparición reciente, afecta preferentemente región epitroclear y zonas extraganglionares.
d) Linfoma de la zona marginal
Es el NHL más común de linfocitos B de baja malignidad, ocupa el segundo lugar en frecuencia. Puede ser esplénico, extraganglionar de MALT o ganglionar.
Constituye casi el 6% de los NHL. Es un linfoma de grado intermedio que a semejanza del NHL indolente no es curable con los tratamientos. La mediana de supervivencia con la enfermedad es de 5-10 años.
Necesita a menudo regímenes de QT más intensivos con o sin trasplante autólogo de células madre para obtener un lapso de respuesta más razonable. R-HiperCVAD (Rituximab, ciclofosfamida, vincristina, doxorrubicina, dexametasona, citarabina y metotrexato).
Hay que progrmar a pacientes que responden a medidas de salvamiento por trasplante de células madre con supervivencia de 30-50% sin enfermedad y a largo plazo.
e) Linfoma linfoplasmacítico
Alrededor de 1% de todos los NHL, que corresponde a linfocitos B de baja malignidad con diferenciación linfoplasmático vinculado a menudo con paraproteína de tipo IgM monoclonal.
El Rituximab en monoterapia puede ser útil.
f) Trastornos de linfocitos T maduros periféricos
Los NHL de linfocitos T son mucho más raros que los de linfocitos B, hay menos conocimiento de su biología y no existe gran adelanto en los tratamiento.
Uno de ellos es la micosis fungoide o linfoma cutáneo de linfocitos T, que es un linfoma de aparición gradual propio de individuos que durante varios años han tenido lesiones eccematosas o dermatitis antes de confirmarse el diagnóstico. Las lesiones cutáneas evolucionan de manera progresiva desde máculas hasta placas para que al final formen tumoraciones cutáneas. El TX es meramente paliativo. Frecuente en hombres, 65 años, raza negra.
El linfoma de linfocitos T no especificados incluye diversas entidades que constituyen el 15% de todos los NHL en adultos. Comprende un conjunto de linfomas heterogéneos que muestran enorme variación en sus signos y no tienen hallazgos típicos de otros subgrupos de PTCL específicos.
El linfoma de linfocitos T angioinmunoblástico comprende el 20% de los NHL de linfocitos T y casi 4% de todos los NHL diagnosticados. El cuadro inicial incluye linfadenopatía, hepatoesplenomegalia, síntomas B, erupciones, poliartritis y anemia hemolítica. Más del 80% busca atención en una etapa avanzada y es común el compromiso de la médula ósea.
El linfoma de células grandes anaplásico ocupa el siguiente lugar después del anterior como el linfoma más común de linfocitos T, pero es más frecuente en niños y constituye hasta el 10% en este grupo de edad. Posee mejor pronóstico que el PTCL, sobre todo si es positiva a ALK que tiene un OS de ocho años de 82% en comparación con 49% con el caso de la enfermedad negativa a ALK.
La leucemia / linfoma de linfocitos T adultos (ATLL) muestra su mayor prevalencia en Japón y países de la cuenca del Caribe. Es una neoplasia que precipita la infección por HTLV-1 y a meno se transmite por la leche de madres infectadas. Su pronóstico varía con mediana de supervivencia de 6, 10 y 24 meses.
El linfoma de linfocitos T/NK extraganglionar de tipo nasal aparece con la infección por VEB en casi todos los casos, es más frecuente en Asia y áreas nativas del Perú. Es un tumor en la zona aerodigestiva. Es más com´n en varones y la mediana edad diagnóstica es de 60 años. El TX de la enfermedad incipiente es una combinación de QT (etopósido, ciclofosfamida, cisplatino, dexametasona) y RT.
4. ¿CÓMO CITO ESTE ARTÍCULO PARA MIS TRABAJOS?
Medina I., Generalidades de Linfomas Hodgkin y No Hodgkin: Repaso ENARM [Internet]. México: Interneuronamed; 2021 jul. [Consultado - Revisado]. Disponible en: https://interneuronamed.blogspot.com/2021/10/generalidades-de-linfomas-hodking-y-no.html
Modificar la sección de "[Consultado - Revisado]" con la fecha de hoy como alguno de los ejemplos siguientes:
1. [Revisado 01 jul 2021]
2. [Consultado 01 jul 2021]

Tienes artículos tan hermosos en tu blog. Permíteme compartir un testimonio sobre cómo me curé del herpes y el vih después de beber Dr. Jekawo a través de la dirección de correo electrónico www.drjekawo.com: drjekawo@gmail.com, un gran herbolario con poder ancestral para curar todas las enfermedades. Me sentí muy devastado cuando me encontré con el Dr. Jekawo después de tanta medicación que no tuvo ningún efecto, pero el Dr. Jekawo preparó medicinas a base de hierbas que luego me enviaron a través del servicio de mensajería de UPS aquí en Texas. Bebí las medicinas a base de hierbas como me indicó el Dr. Jekawo y hoy estoy completamente curada. También recomiendo al Dr. Jekawo a mis familiares y amigos que padecen diferentes enfermedades como VPH, diabetes, herpes y EPOC. Todos recibieron su tratamiento a base de hierbas y se recuperaron por completo. El Dr. Jekawo cura todas las enfermedades. Visite su sitio web si está enfermo.
ResponderEliminar